While the past few years of our life has been much dictated by COVID-19, Alzheimer’s disease (AD) is the twenty-first-century plague. Every three seconds, an individual is diagnosed with dementia. Over 55 million people worldwide were living with AD in 2020, and this number will almost double every 20 years, reaching over 100 million in 2050. Furthermore, dementia has significant social and financial implications: the total estimated annual worldwide cost of dementia is approximately US$ 1 trillion, and this figure is forecast to rise to US$ 2.8 trillion by the end of this decade1. Despite its importance, the exact causes of Alzheimer's disease are still not fully understood after hundred years of research. But what we do know so far is healthy neurons are supported internally by structures called microtubules, which help guide nutrients and molecules from the cell body to the axon and dendrites. Tau proteins normally bind to and stabilize microtubule structure. In Alzheimer’s disease, however, tau proteins detach from microtubules and reorganize themselves into structures called neurofibrillary tangles that have become the pathological hallmark2. In AD brain not only the number but also the spreading of tau tangles correlates to the severity of dementia symptoms3. Yet, monomeric tau is highly soluble and intrinsically disordered such that tau shows little tendency for aggregation. While the exact underlying mechanism is still unclear, recent studies suggest post-translational modifications in the form of excessive phosphorylation can trigger the formation and therefore the growth of tau aggregates observed in AD brain2. Furthermore, increasing evidence suggests that hyperphosphorylated tau can lead to microtubule dysfunction and impair axonal transport of organelles, altogether resulting in synaptic dysfunction4,5.
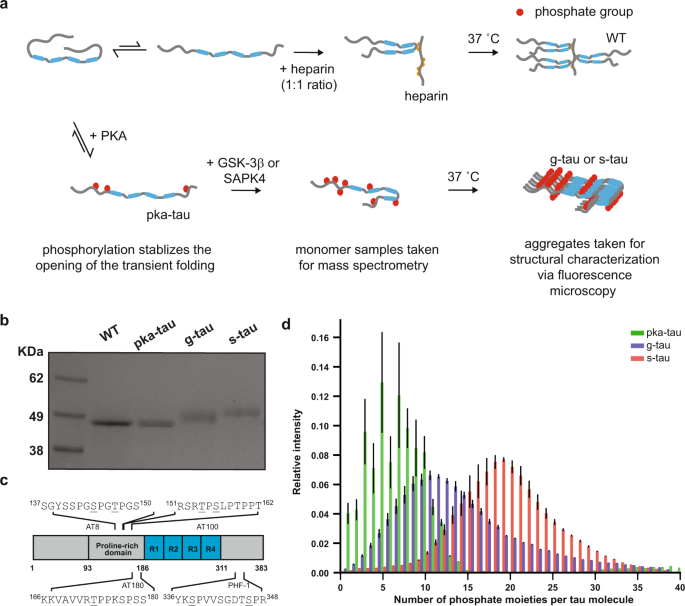
The identification of phosphorylation sites responsible for neurotoxicity remains elusive, and the challenge mainly lies in the heterogeneous and combinatorial nature of tau phosphorylation. Therefore, to examine the pathological consequences of tau (hyper)phosphorylation, we needed to be able to reproducibly generate disease-relevant phospho-tau in quantities that meet the demand for subsequent structural and toxicity characterization studies. In our current study, we modified a previously published protocol and sequentially hyperphosphorylated tau at AD-specific epitopes in vitro, as measured by our high-resolution native mass spectrometry and liquid chromatography-tandem mass spectrometry. By comparing with previous reports on AD-derived PHF tau and other (hyper)phosphorylated tau synthesized in vitro, our hyperphosphorylated tau were the most disease-relevant species in terms of their phosphorylation sites6.
We then employed fluorescence microscopy and transmission electron microscopy to characterize the effects of hyperphosphorylation on the aggregation process and morphology of tau without the use of any external inducer. It is important to note that external inducers such as heparin and other polyanionic molecules have commonly been used to induce aggregation in vitro since monomeric tau shows little intrinsic tendency for aggregation in vitro. Yet, recent technological breakthroughs and developments of more sensitive biochemical assays suggested that heparin-induced tau and AD-derived tau have distinct properties7. As these observations called the use of external inducers into question, our current study focused on the impact of tau phosphorylation on its susceptibility to aggregation and our results showed that hyperphosphorylation could trigger tau to spontaneously assemble into small amorphous aggregates at physiological concentrations.
Following our structural investigation, we then evaluated whether such pronounced morphological differences might translate to a gain of toxic function for tau. To address this question, a high-throughput calcium assay developed by our lab was used to measure the ability of tau aggregates to disrupt lipid membrane integrity as a proxy of tau cytotoxicity. We found that these hyperphosphorylated tau aggregates could better cause membrane permeabilization than WT aggregates made using an inducer. Furthermore, owing to the direct involvement of ab amyloid and tau tangles in AD pathology, recent studies have also highlighted a central role of neuroinflammation in the pathophysiology of AD. Microglia aggregate around plaques and clear them by phagocytosis. However, inefficient processing of such protein aggregates would lead to further seeding events and trigger prolonged expression of proinflammatory cytokines which could in turn cause more protein aggregation in healthy neurons and recruit monocytes, T-cells, and monocyte-derived macrophages which will remove synapses by phagocytosis8,9. Thus, to better characterize the toxicity profile of hyperphosphorylated tau, we used monocyte-derived macrophage cells as our model cells to investigate how distinct species of tau aggregates may affect immune responses. Our results revealed that amorphous hyperphosphorylated tau aggregates could induce temporary calcium transients, elicit oxidative stress, and promote the production of inflammatory species via the Toll-like receptor 4 signaling pathway. Our work, therefore, provides a potential mechanistic insight connecting tau aggregation due to hyperphosphorylation to neuroinflammation and neurodegeneration, thereby offering a possible molecular basis for AD and other tauopathies.
References
- C., P. World Alzheimer report 2018: the state of the art of dementia research. New frontiers (2018).
- Alonso, A. D. C., Zaidi, T., Novak, M., Grundke-Iqbal, I. & Iqbal, K. Hyperphosphorylation induces self-assembly of into tangles of paired helical filamentsstraight filaments. (2001).
- Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T. & Hyman, B. T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639 (1992).
- Cai, Q. & Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. Journal of Alzheimer’s Disease vol. 57 1087–1103 Preprint at https://doi.org/10.3233/JAD-160726 (2017).
- Ittner, L. M. & Götz, J. Amyloid-β and tau - A toxic pas de deux in Alzheimer’s disease. Nature Reviews Neuroscience 12, 67–72 (2011).
- Hanger, D. P. et al. Novel phosphorylation sites in Tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. Journal of Biological Chemistry 282, 23645–23654 (2007).
- Zhang, W. et al. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 8, e43584 (2019).
- Becher, B., Spath, S. & Goverman, J. Cytokine networks in neuroinflammation. Nature Reviews Immunologyvol. 17 49–59 Preprint at https://doi.org/10.1038/nri.2016.123 (2017).
- Hopp, S. C. et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. Journal of Neuroinflammation 15, 269 (2018).
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in