
Every individual inherits one copy of the human genome from each of their parents. Analyzing an individual’s genome can help understand their risks for disease, and to understand who their ancestors were and where they lived stretching back across generations. Genome information can be determined by sequencing, which is comprehensive but also expensive, or by genotyping chips that affordably measure specific positions in the genome known to vary across populations.
After genotyping, a method called imputation is often used to predict a much denser set of positions across the entire genome by using a reference panel of haplotypes (derived from sequencing) and leveraging linkage disequilibrium (the correlation between alleles across individuals). However, because historical genome studies have substantially over-represented individuals of European ancestry and under-represented individuals of other and mixed ancestries, reference panels are less accurate for these under-represented ancestries. As a result, many analyses such as ancestry inference and the discovery of genetic factors for disease risk are less powerful for non-European ancestries than they would be with more equitable representation across ancestries.
In order to help improve the usefulness of genomic analysis for individuals of African American ancestry, in our manuscript “A population specific reference panel for improved genotype imputation in African Americans”, we develop an expanded reference panel using a combination of additional genome sequencing and developed new computational methods to increase the accuracy of the resource.
We sequenced 2,269 23andMe customers who had consented to participate in research and had high amounts of genetically-inferred sub-Saharan African ancestry. All individuals were re-contacted and consented to having their individual-level sequencing data made available to qualified researchers.

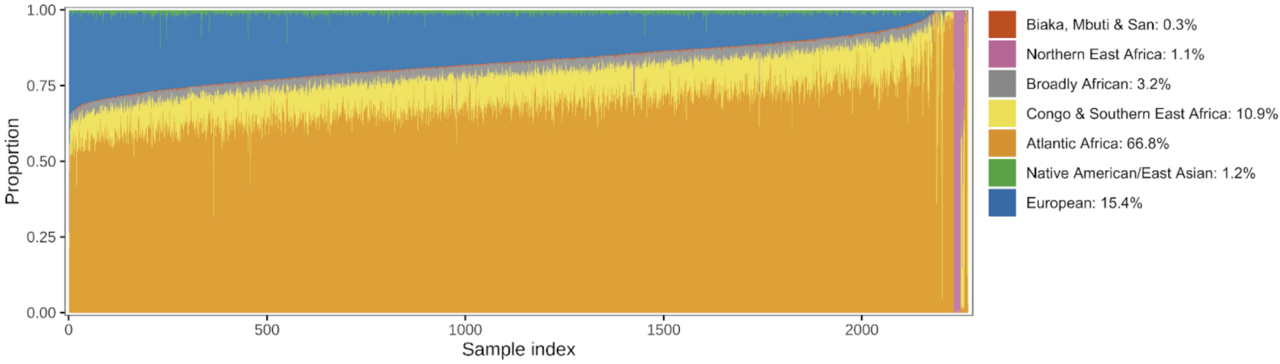
Figure 1. The ancestry composition of our reference panel. Estimated ancestry proportions for each of the 2,269 sequenced individuals in the AFAM panel. Only African regions and other regions that contributed substantially to admixture were included. Each column represents an individual colored by their respective estimated ancestry.
In order to improve the accuracy of the reference panel, we evaluated the potential of new informatics methods for variant discovery in sequencing data, specifically the combination of DeepVariant for single sample variant calling and GLnexus for combining calls across a cohort. We found that this combination enabled higher accuracy of sequence analysis, as measured by concordance with high-quality genotype calls, across 292 samples.
 |
Figure 2. F1 metric (harmonic mean of precision and recall; higher is better) per sample for SNPs as a function of sequence coverage in a subset of 23andMe AFAM samples (N=292). Each sample produces three points at a single coverage level, indicating the F1 performance of that sample using each of the three variant callers. DeepVariant substantially outperforms both versions of GATK, particularly on lower coverage data. |
After calling variants across all samples in the cohort, we began the process of creating and evaluating the reference panel. We optimized a cohort-level variant filtering approach which used the properties of calls at each position across the cohort to identify and remove potential false positive sites. We experimented with various tools and parameters for building the reference panel, ultimately arriving at an optimized set of parameters for SHAPEIT 4.
We compared the performance of our imputation panel against four other widely-used panels by imputing 103 African Americans with truth data available. We show that the reference panel generated for this paper outperformed three of the panels (1KGP, HRC and CAAPA). Although the much larger TOPMed imputation panel was the most accurate, it cannot be readily downloaded.

Figure 3. Imputation performance for five different panels using a truth set containing 103 GTEx WGS individuals imputed with an emulated 523K 23andMe microarray. Aggregate R2 between the imputed dosages and sequence genotypes as a function of the alternate allele frequency reported for African Americans in the gnomAD r3 dataset. We treat variants missing from the panel to be imputed as homozygous reference here, which penalizes panels that have missing variation.
We have fully released our reference panels and raw sequencing data for others to either use directly, or to combine the reference panel with their own panels. It is our hope that by releasing the panel, and by describing the optimized manner of generating accurate reference panels for many samples at lower coverages, we can contribute to improving genomic resources and the utility of genomic analysis for individuals of African ancestry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in